药物代谢动力学(Pharmacokinetics),简称动力学,是机体对药物的作用,包括药物的吸收(Absorption)、分布(Distribution)、代谢(Metabolism)和排泄(Excretion),即所谓的ADME。药物代谢动力学通过应用动力学的原理与数学处理方法,定量地研究药物(包括外来化学物质)通过各种给药途径进入机体后的吸收、分布、代谢和排泄等过程的动态变化规律,即研究给药后药物在体内的位置、数量、疗效与时间之间的关系,并提出解释这些关系所需要的数学关系式的科学。药物在进入体内后,经过吸收入血液,并随血液透过生物膜进入靶组织与受体结合,从而产生药理作用,部分药物还在肝脏和肾脏等组织中发生代谢,进而排出体外。药物在体内的过程可用图1描述。
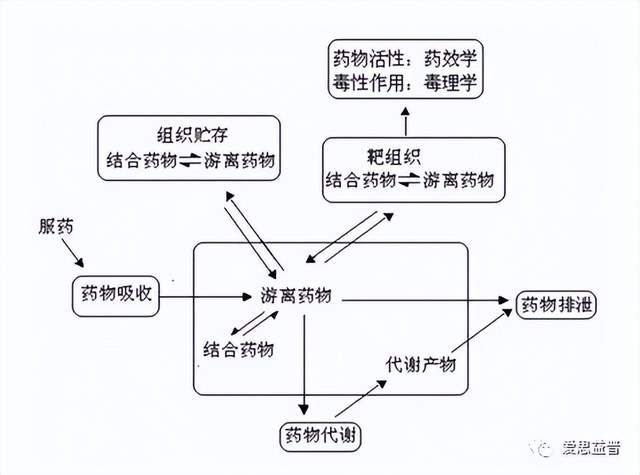
图1. 药物在体内过程
药物吸收是指药物从给药部位进入血液循环的过程,除动脉和静脉给药之外,其他给药途径均存在吸收过程,但是不同的给药途径,如口服、皮下、透皮、静脉和肌注,药物吸收的比例是不同的,这主要是因为药物在吸收过程中,会受到很多因素的影响。因为口服给药方便、经济,即可免除创伤性给药的不适,又能增加患者的依从性,所以口服途径是首选的给药途径,因此本文主要说明口服给药后的胃肠道药物吸收。
要研究药物是否被吸收,首先要确定药物的理化性质,如溶解度、pH值和LogD值。大多数药物以离子化或非离子化的形式存在弱酸或弱碱溶液中。离子化的药物是亲水的,不能穿过细胞膜;而非离子化的药物是相对亲脂性较强的,可以通过简单的扩散穿过细胞膜。药物的分布是由膜两侧的pH梯度和药物的pKa决定的,弱酸性药物在低pH值的介质中容易被吸收,如在胃中;而弱碱性药物则需要在到达pH值较高的小肠时才会被吸收。药物的溶解度实验和LogD值检测实验能够确定药物的溶液中的溶解性和亲脂性,能够很好的为确定药物是否易被吸收提供依据。
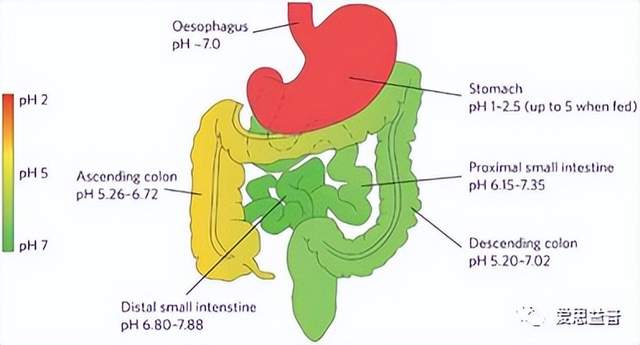
图2. 胃肠道酸碱环境

图3. 动力学溶解度和热力学溶解度实验流程
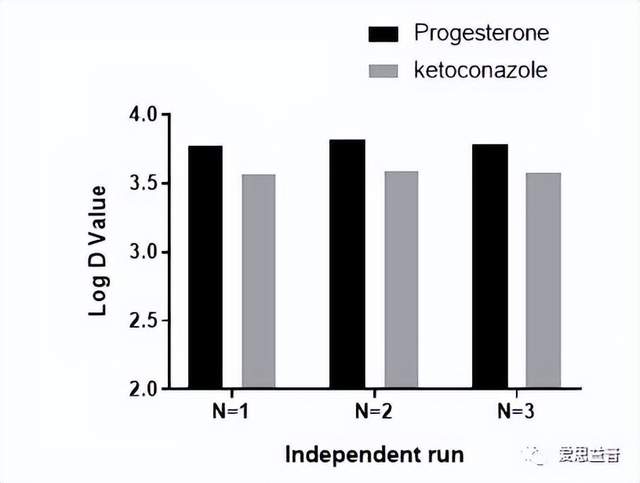
图4. 亲脂性LogD pH 7.4数据
其次,药物粒径大小和表面积、溶解率、无定形性、多形性特征和剂型的性质也会影响系统药物的吸收。溶解率是指在pH值、溶剂成分和合适温度的标准条件下,在表面积不变的情况下,每次变成溶液的固体物质的数量。而颗粒的大小与溶解率成反比,因此减小颗粒尺寸可以增加表面积,从而提高溶解率,如将药物颗粒微粉化可提高溶解率和溶解度。对于临床前研发的药物来说,一般药物形态多是粉末,而给药的剂型静脉注射需要是澄清的溶液,其他口服、腹腔等给药方式可以是澄清或者混悬液。对于难溶药物来说,一般先用DMSO等强助溶剂溶解,后选用生理盐水等溶液进行稀释到所需浓度,如果在试药过程中不溶,则通过用缓冲盐溶液调节pH值(2~9)观察是否溶解,如果还不溶,则需要再加入助溶剂或者表面活性剂进行溶解,一般情况下难溶药物都会得到很好的改善,对于特殊药物就需要再进行溶媒开发。
奥拉帕尼(Olaparib)是聚二磷酸腺苷核糖聚合酶(PARP)抑制剂,PARP酶涉及正常细胞动态平衡,如DNA的转录,细胞周期调节和DNA修复。我们通过尾静脉注射和口服给药研究其在小鼠体内的药代动力学变化规律,发现1)重复尾静脉给药相同剂量、相同溶媒,其重复性较好,动物批次的变化对其影响较小;2)改变口服的溶媒(从澄清溶液变为混悬液),重复口服给药相同剂量、不同溶媒,明显可以看出药物的达峰时间后移,由之前的0.14 h 变为0.28 h,达峰浓度明显降低,药时曲线下面积和生物利用度也明显降低,平均滞留时间延长,但是对药物的半衰期影响较小,说明药物的溶媒改变可以明显影响药物的吸收,进而影响药物在体内的代谢和排泄。

图5. Olaparib在雄性小鼠体内的药代动力学研究(A. PO-澄清溶液;B. PO-混悬溶液)

第三,当药物到达小肠后(一般为弱碱性药物),必须穿过小肠上皮细胞膜才能到达全身循环,其主要转运系统包括被动(简单)扩散和载体介导的转运系统。其中被动扩散是药物最常见的吸收机制,药物分子可根据浓度梯度从较高的药物浓度向较低的浓度移动,直到达到平衡。被动扩散可以发生在水性或脂质环境中,其中在水性环境中被动扩散主要发生在身体的水性隔室中,例如血管内皮中的水性孔,其可以阻挡与白蛋白或其他大的血浆蛋白结合的药物。另一方面,脂质扩散是通过体内的脂质区发生的,体内有很多的脂质屏障将身体各部分分开,因此脂质扩散被认为是药物渗透性的最重要因素。其次,载体介导的转运系统,主要包括主动转运和易化扩散,两者的主要区别是,主动转运是需要消耗能量的,能将药物由低浓度向高浓度的一侧转运,而易化扩散不需要消耗能量,是顺梯度将药物从浓度高的一侧转运至浓度低的一侧。两者的相同点是在浓度达到一定程度后,载体上的结合位点可能会达到饱和,之后剂量的增加不会影响药物的浓度,另外如果出现对载体选择性更强的药物时,会出现竞争抑制现象。虽然有些转运体有利于吸收,但其他转运体如P-糖蛋白(P-gp)可有效阻碍药物吸收,P-gp(也被称为MDR1)是一种能量依赖性的外排转运体,促进药物外排回肠腔,从而限制整体吸收。
体内实验评价药物在小肠内是否吸收的主要指标包括药物的口服生物利用度、口服药物的达峰时间和达峰浓度。其中口服生物利用度一般为绝对生物利用度,将口服药物的AUC与静脉给药后的AUC(如果非相同剂量给药,需进行剂量校正)进行比较,可以确定口服药物的吸收特性。体外实验则可以通过测定药物的Log D值、建立Caco2模型、Transporter和PAMPA等手段进行检测,从而表明药物的部分药代特性,对新药研发过程中先导化合物的筛选起到关键性的作用。
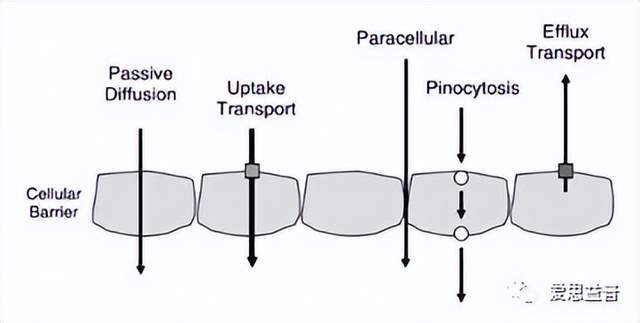
图6. 常见转运类型
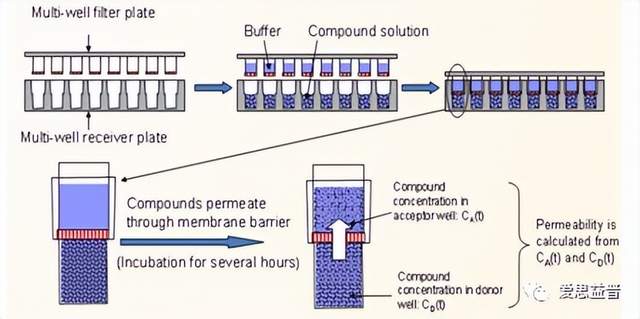
图7. PAMPA平行人工膜渗透模型实验流程
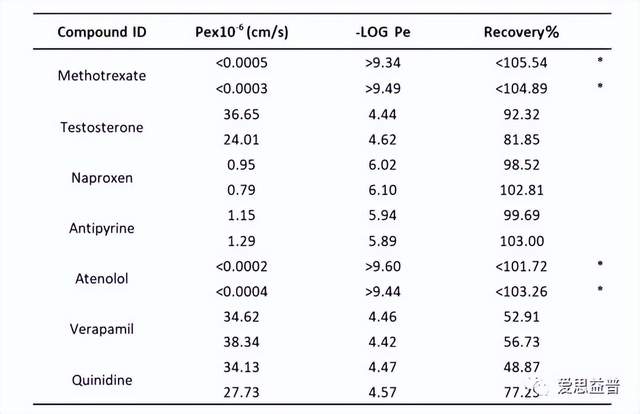
图8. PAMPA平行人工膜渗透模型数据
*1 was used to calculate Pe if CA(t) was BLOQ.
Test Concentration: 10 uM, Incubation time: 16 hours
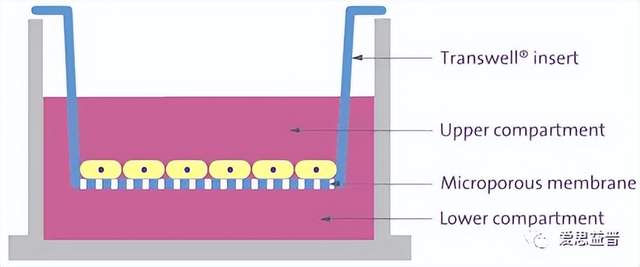
图片
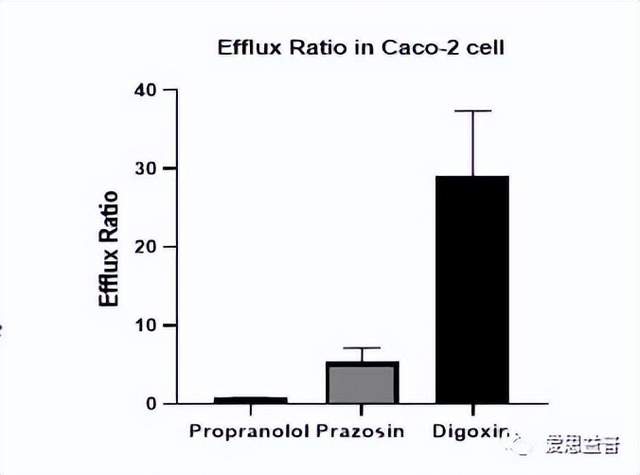
图9. Caco-2模型及数据
爱思益普DMPK团队,建立了系列和药物吸收相关的体外和体内实验,包括药物溶解度实验、Log D值实验、Transporter和PAMPA等体外实验,还包括大小鼠及犬的静脉注射、口服给药、腹腔注射和皮下注射等体内实验,能够提供定制化的体内外实验,很好的为客户提供药代动力学服务。
结合爱思益普强大的靶点体外方法开发及筛选的能力,完善的激酶谱学筛选、脱靶效应评价、细胞谱学筛选等平台,可以为客户提供更完整的药物筛选和评价策略,加速中国新药研发。
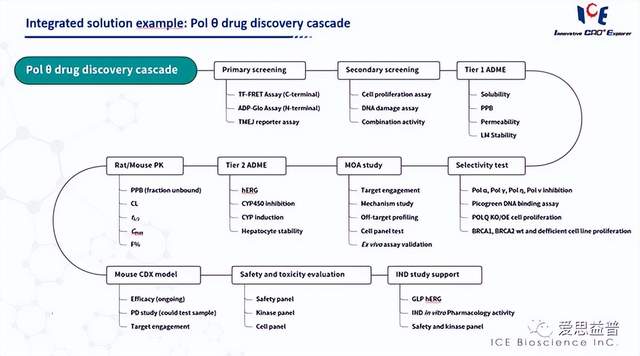
欢迎垂询,获得POLQ筛选平台技术资料。
[1] 王广基, 柳晓泉, 刘晓东. 药物代谢动力学[J]. 化学工业出版社, 2005.
[2] Abdulrahman A. Alagga 1, Vikas Gupta. Drug Absorption. Book. 2021 Aug 29.
[3] Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002 Jun;42(6):620-43. - PubMed
[4] Stoll L, Gentile L. Linking tricyclic antidepressants to ionotropic glutamate receptors. Biochem Biophys Res Commun. 2005 Jul 29;333(2):622-7. - PubMed
[5] Di, L., et al. (2003). "High throughput artificial membrane permeability assay for blood-brain barrier." Eur J Med Chem 38(3): 223-232.